|
|
|
|
UVOD
Pre više od četiri decenije mnoge zemlje su pokrenule programe
neonatalnog skrininga u cilju otkrivanja novorođenčadi sa naslednim
metaboličkim i endokrinološkim oboljenjima za koja bi rana
dijagnostika i lečenje sprečila ozbiljne i trajne poremećaje
zdravlja. Fenilketonurija je u mnogim zemljama bila prvi poremećaj
uvršten u novorođenački skrining. U decenijama nakon toga program se
širio postepeno, i obuhvatao sve veći broj teških poremećaja koji za
posledicu imaju visok stepen fizičkog i intelektualnog invaliditeta.
Svetska zdravstvena organizacija definiše ulogu skrininga kao
otkrivanje bolesti koja se može lečiti, sa adekvatno shvaćenom
prirodnom istorijom, u asimptomatskoj fazi, kako bi se započelo
lečenje i sprečili simptomi ili da bi se odložile komplikacije.
Skrininig novorođenčeta se počeo primenjivati 1960. godine radom
američkog mikrobiologa Dr Roberta Gatrija (Robert Guthrie). Prva
interncionalna diskusija o skriningu novorođenčeta pod organizacijom
Svetske zdravstvene organizacije održana je 1967. godine kada je
grupa naučnika za kongenitalne poremećaje metabolizma raspravljala o
tehničkim i etičkim aspektima skrininga.
Gatrijev test (Guthrie test) je obavezna mera zdravstvene zaštite i
radi se svakom novorođenčetu, bilo da je ono zdravo ili bolesno,
rođeno u ili pre termina. Ova laboratorijska analiza se uglavnom
izvodi već u porodilištu, najčešće od 48 do 72 sata od rođenja
novorođenčeta, mada može da se radi i do 8. dana života
novorđenčeta. Važećem preporukom Savetodavnog komiteta za nasledne
bolesti kod novorođenčadi i dece, čija aktuelna verzija datira iz
2016. godine u SAD je definisan "preporučeni univerzalni skrinig
panel" koji se sastoji od osnovnog spiska od 34 oboljenja i
proširenog spiska na kojem se nalazi jos 26 bolesti. Oboljenja za
koja se preporučuju skrining mogu se klasifikovati na nekoliko
grupa: poremećaje metabolizma organskih kiselina, poremećaj
oksidacije masnih kiselina, poremećaje metabolizma aminokiselina,
endokrine poremećaje i hemoglobinopatije. Od endokrinih poremećaja
skrining se preporučuje na kongenitalni hipotireoidizam i
kongenitalnu adrenalnu hiperplaziju i to u okviru osnovnog panela
[1]. Lista bolesti koje će obuhvatiti skrining test, zavisi od
zdravstvenog sistema države i njenog skrining programa. Koja će se
bolest proveravati najviše zavisi od njene učestalosti, od
dostupnosti terapije ali i od toga koliko je zemlja razvijena i ima
li sredstva da plati skrining za svu novorođenčad.
U Crnoj Gori od 2007. godine kao obavezni vid zdravstvene zaštite
novorođenčeta uveden je neonatalni skrining na hipotireozu, i jedina
je bolest iz grupe naslednih endokrinoloških oboljenja koju skrining
obuhvata. Od zemalja u okruženju, Slovenija ima najbolji skrinig
program gde je pored hipotireoze i fenilketonurije uvršteno još
sedamnaest oboljenja. Hrvatska ima skrining program koji obuhvata
osam bolesti: fenilketonuriju, hipotireozu, tri poremećaja
razgradnje masnih kiselina, glutarnu aciduriju tipa 1, izovaleričnu
acidemiju, nedostatak karnitina.
Novorođenački skrining na fenilketonuriju
Skrining na fenilketonuriju je preduslov za ranu primenu dijete,
koja je neophodna za prevenciju teških neuroloških poremećaja kod
dece sa dijagnostkovanim oboljenjem.
Fenilketonurija je najčešći urođeni metabolički poremećaj koji
uzrokuje težak stepen fizikčkog i pshičkog invaliditeta ukoliko se
blagovremeno ne dijagnostikuje i ne započne terapijski tretman.
Fenilketonurija je bolest koja se može lečiti i navedena je u
nacionalnom programu skrininga novorođenčadi u zemljama širom sveta.
Novorođenčad sa pozitivnim indikacijama skrininga mogu postići
zadovoljavajući terapijski efekat blagovremenom kontrolom unosa
fenilalanina nakon postavljanja dijagnoze. Kombinacija rane
dijagnoze i početka lečenja rezultira normalim telesnim i
intelektualnim razvojem za većinu dece sa fenilketonurijom.
Fenilketonurija i druge hiperfenilalaninemije su skupina naslednih
poremećaja koje nastaju zbog poremećaja u oksidaciji aminokiseline
fenilalanin u tirozin [2]. Fenilketonuriji pripada posebno mesto
među naslednim metaboličkim bolestima. To je prva bolest iz te
skupine u kojoj je jasno utvrđena veza između naslednog biohemijskog
poremećaja i mentalne zaostalosti (Asbjorn Fӧlling 1934.), prva
bolest iz te kategorije za koju je otkrivena mogućnost lečenja
dijetom (Horst Bickel 1954.) i prva za koju je izrađen
labaratorijski test koji se upotrebljava u skriningu novorođenčadi u
celokupnoj novorođenačkoj populaciji (Robert Guthrie 1963) [3].
Prevalenca fenilketonurije u svetu se kreće oko 1: 10 000
novorođenčadi [4].
Fenilalanin je esencijalna aminokiselina, od koje se nakon
resorpcije iz creva manja količina ugrađuje u telesne proteine, a
preostali, veći deo mora se uz pomoć enzima fenilalanin-hidroksilaze
u jetri oksidirati u tirozin. Uzrok fenilketonurije su mutacije gena
koji se nalazi na hromozomu 12 koji kodira jetreni enzim
fenilalanin-hidroksilazu. Posledica je insuficijencija enzima i
nemogućnost oksidacije fenilalanina u tirozin sa povećanjem
koncentracije fenilalanina i njegovih "nenormalnih "metabolita u
ćelijama i telesnim tečnostima. Danas još nije poznat mehanizam
kojim fenilalanin ili njegovi metaboliti u velikim koncentacijama
oštećuju funkciju mozga, ali je činjenica da njihovo održavanje u
normalnim granicama kod dece sa fenilketonurijom odgovarajućim
dijetalnim režimom sprečava oštećenje mozga [5].
Deca sa klasičnom fenilketonurijom u prvim danima i nedeljama života
nemaju uočljivih simptoma. Tek nakon nekoliko nedelja javlju se
znaci usporenog psihomotornog razvoja, deca ne nauče hodati, sedeti
u pravo vreme, 25 % dece ima epileptičke napade, razvija se
hipotonija muskulature, psihomotorni nemir, promene ponašanja,
mikrocefalija, zaostatak u telesnom razvoju. Oko četvrtine zahvaćene
dece ima dojenački ekcem, hipopigmentaciju kože i kose, miris znoja
i mokraće na miševe što potiče od fenilmlečne kiseline koju ta deca
izlučuju. Već tokom prve godine dolazi do teške mentalne retardacije
(IQ 30) [6].
Slika 1. Dete sa fenilketonurijom
Izvor: https://img.medscapestatic.com/pi/meds/ckb/07/44107tn.jpg
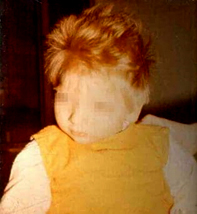
Kako se kod svakog novorođenčeta radi skrinig na fenilketonuriju
(Guthrie test), u dece sa pozitivnim Guthrie skrinig testom određuje
se se koncentracija fenil-alanina i tirozina u krvi. Na osnovu
vrednosti fenilalanina u krvi, bolest se klasifikuje kao blaga
hiperfenilalaninemija: 120–360 mmol; blaga siva zona 360–600 mmol;
blagi oblik fenilketonurije: 600–900 mmol; umereni: 900–1200 mmol i
klasični >1.200 mmol [7].
Lečenje fenilketonurije se sprovodi doživotnim ograničenjem unosa
fenil-alanina do količine neophodne za izgradnju vlastitih proteina
od rođenja. U odojčadi se isključivo koriste mlečne formule sa malo
fenil-alanina. Primena dijete ima trostruki cilj:
1.Sprečva se akumulacija prekomerne količine fenilalanina u krvi (a
samim tim i u mozgu) strogom kontrolom prirodnog unosa
proteina/fenilalanina.
2.Zamena prirodnog proteina koji je uklonjen iz ishrane bezbednim
proteinom ili proteinom bez fenilalanina, koji se naziva sintetički
protein, smeša/suplement aminokiselina ili zamena za proteine. Sve
zamene za proteine su bez fenilalanina ili imaju veoma malo
fenilalanina.
3.Postizanje normalnog rasta i statusa uhranjenosti. Ovo se postiže
osiguravanjem da ishrana sadrži izbalansiran unos svih hranljivih
materija i energije. Suplementi vitamina i minerala se ili dodaju
zameni proteina ili daju kao poseban dodatak.
U ishrani se doživotno ograničava unos namirnica koje obiluju
fenilalaninom: mleko, mlečni proizvodi, meso, riba, piletina, jaja,
pasulj, orasi. U ishrani se preporučuje unos voća, povrća, žitarica
[8].
Prognoza nelečene fenilketonurije je loša sa obzirom na propadanje
mentalnih i nervnih funkcija, propratnu simptomatsku epilepsiju i
teškoće i komplikacije koje prete takovom detetu. Oko polovine
nelečene dece doživi 20 godina, oko trećine 30 godina. Uz
blagovremenu dijagnostiku u najranijoj dobi i adekvatnu ishranu deca
sa lečenom fenilketonurijom se ne razlikuju od zdravih vršnjaka.
Prevencija fenilketonurične embriopatije započinje pre rađanja
deteta, kada gravidna žena koja ima fenilketonuriju sprovodi dijetu
bez fenilalanina. Ako pre koncepcije i u toku trudnoće dijeta nije
stroga doći će do oštećenja centralnog nervnog sistema fetusa,
urođenih srčanih mana i mikrocefalije. Po rođenju novorođenčetu se
radi Gatrijev test (Guthrie test).
Uzorak treba uzeti svakom zdravom, bolesnom, donešenom i nedonešenom
novorođenčetu. Tačan period za uzimanje uzoraka ne bi trebalo da
bude kraći od 48 sati hranjenja proteinima i ne bi trebalo da
prelazi 30 dana od rođenja; međutim, idealan period bi bio između
trećeg i sedmog dana rođenja kod novorođenčadi [9].
Budući da antibiotska terapija može test na fenilketonuriju učiniti
lažno negativnim uzorak se uzima u načelu nakon završetka
antibiotske terapije. Najsigurnije mesto za uzimanje uzorka krvi je
dorzalna strana pete novorođenčeta. Označeni krug mora biti u
potpunosti ispunjen krvlju, ne smeta ukoliko je krv prešla rubove
kruga. Pre uboda deteta treba sačekati da se da se dezinfekciono
sredstvo kojim je koža obrisana potpuno osuši. U suprotnom sa
uzorkom krvi se meša dezinfekciono sredstvo te je takav uzorka
neupotrebljiv. Jod i sredstva koja sadrže jod se ne upotrebljavaju
jer ometaju određivanje tireotropina za dijagnostikovanje
kongenitalne hipotireoze. Na poleđini papira važno je napisati da li
dete uzima antibiotike i je li teško bolesno.
Novorođenački skrinig na hipotireozu
Kongenitalna hipotireoza može se dijagnostikovati kasno ili može
proći potpuno nedijagnostikovano, izazivajući poremećaje zdravlja
deteta, ekonomski i socijalni teret za porodicu. Terapijski tretman
dijagnostikovane kongenitalne hipotireoza je jednostavan, jeftin i
efikasan. Sa ranom dijagnozom i terapijom novorođenče se razvija
normalno bez mentalnog hendikepa i postaje produktivan član društva.
Patnja deteta, postojanje ekonomskog i socijalnog tereta uzrokovanih
kongenitalnom hipotireozom, obavezala je institucije mnogih zemalja
da novorođenački skrining na hipotireozu uvrste u obavezan vid
zdravstvene zaštite deteta.
U Crnoj Gori, kao obavezan vid zdravstvene zaštite djeteta uveden je
skrining na hipotireozu 2007. g. Do danas, kongenitalna hipotireoza
je jedino endokrino oboljenje obuhvaćeno skrining programom
novorođenčadi.
Glavne kliničke karakteristike nelečene kongenitalne hipotireoze su
poremećaj rasta i odloženi neurokognitivni razvoj koji rezultira
mentalniom retardacijom.
Širom sveta stopa incidence kongenitalne hipotireoze je 1: 2000-4000
novorođenčadi, dok za područja koja su deficitarna jodom beleže veću
stopu incidencije [10].
Slika 2. Klinička slika kongenitalne hipotireoza
Izvor:https://www.researchgate.net/publication/44662677/
figure/fig4/AS:279090520182836@1443551773718/Infant-with-congenital-hypothyroidism-
A-3-month-old-infant-with-untreated-CH-picture_Q320.jpg
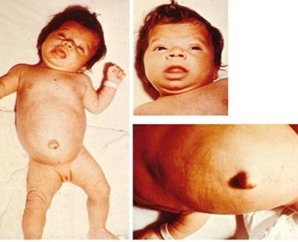
Kongenitalna hipotireoza se dijagnostikuje na rođenju pomoću
Gatrijevog testa (Guthrie test). Ovaj test se bazira na merenju
vrijednosti (TSH) tireostimulišućeg hormona ili (T4) tiroksina. Ako
je nivo T4 u krvi iz uboda u petu nizak a povišen TSH rezultati
skrininga ukazuju na postajanje kongenitalne hipotireoze. Potvrda
dijagnoze se postavlja analizom hormona iz venske krvi gde se takođe
meri nivo TSH i T4. Ako je vrednost T4 hormona niska, a vrednost TSH
povišena dijagnoza je defiitivno potvrđena [11].
Cilj supstitucione hormonske terapije je dovesti dete u stanje
eutireoze. Kod dijagnostifikovane kongenitalne hipotireoze terapija
se započinje sa punom dozom hormona kako bi se sprečili ili umanjili
štetni efekti hipotireoze na razvoj centralnog nervnog sistema.
Preporučuje se održavanje T3 i T4 na gornjoj granici normale.
Početkom terapije normalizuje se nivo T4 i T3 i dolazi do supresije
povišenog TSH. Uz dobro vođenu terapiju postiže se normalan rast i
gube se klinički znaci hipotireoze, ali prognoza mentalnog razvoja
nije tako povoljna i zavisi pre svega od vremena kada je terapija
započeta. Levotiroksin je hormonski preparat koji se koristi u vidu
tableta ili rastvora. Tabletu je potrebno izmrviti i pomešati sa 30
ml tečnosti (vode, mleka ili formule). Rastvor se detetu daje preko
šprica ili pipete, ne treba ga mešati u celokupni obrok u flašici
jer se može desiti da beba ne pojede čitav obrok i da se ne unese
potpuna doza leka. Tokom hormonske terapije neophodno je pratiti
stanje deteta, jer usled predoziranja levotiroksinom mogu se razviti
simptomi hipertireoze: nemir, blage dijareje, sporo napredovanje u
telesnoj težini, nesanica, ubrzan rast.
Usled nedovoljne terapijske doze kod deteta se mogu razviti
letargija, opstipacija, hladni ekstremiteti, neočekivano dobijanje u
telesnoj težini i usporen rast.
Nakon započinjanja hormonske terapije neophodno je pratiti vrednosti
tireoidnih hormona. U prvim mesecima hormonski status se proverava
svakih par nedelja, odnosno na svakih tri do šest meseci tokom
detinjstva, odnosno na svakih 6 do 12 meseci u adultnom dobu [12].
Veliki broj zemalja uvrstio je i hipotireozu u svoj program
novorođenačkog skrininga i to na taj način što se iz istog uzorka
krvi sa filter papira koji se uzima radi traganja za
fenilketonurijom određuje radioimunološki T4 ili TSH.
Novorođenački skrining na galaktozemiju
Zbog nedostatka galaktoza-1-fosfo-uridil-transferaze nastaje
klasična galaktozemija [13]. Usled neaktivnosti ove transferaze,
dolazi do nagomilavanja galaktozo-1-fosfata u jetri, eritrocitima,
slezini, očnom sočivu, bubrezima, srčanom mišiću i moždanoj kori, a
u krvi postoji galaktozemija. Sem intracelularnog nagomilavanja
galaktoze i galaktozo-1-fosfata nalazi se i veća količina
galaktitola. Nakon nekoliko dana hranjenja majčinim mlekom ili
mlečnom formulom koja sadrži laktozu novorođenče postaje anoreksično
i požuti. Novorođenče sa klasičnom galaktozemijom često odbija
hranu, ne napreduje ili gubi na telesnoj masi, povraća nakon obroka,
ima proliv, žuticu, ascites, edeme, hepatomegaliju, letargična je i
hipotonična. Oštećenje jetre može napredovati do fulminantog
zatajenja s encefalopatijom i hemoragijskom dijatezom, a moguće je
zatajenje bubrega [14].
Slika 3. Dete oboljelo od galaktozemije
Izvor:
https://encrypted-tbn0.gstatic.com/images?q=tbn:ANd9GcTpVTHhntyHltIfN9_
IwAGV4X8QUKZkDzQ51mKrGQqKsz5XitFfyvnvkKHrwiQSg4ZNKxA&usqp=CAU
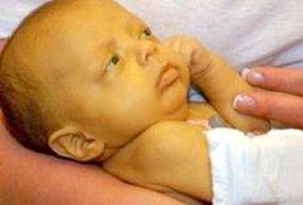
Deca ostaju niskog rasta uz govorne nedostatke kao i poremećaj
držanja tela i ravnoteže tokom adolescencije. Nagomilavanje
galaktoze i galaktitola u očnom sočivu dovodi do brzog formiranja
katarakte, zamućenja očnog sočiva i gubitka vida. Bolest može biti
praćena osteomalacijom, privremenim zatajanjem jajnika, dok teži
oblici galaktozemije su praćeni gubitkom sluha [15]. Lečenje
galaktozemije se zasniva na dijeti bez imalo galaktoze (za dojenčad
je to sojino mleko umjesto kravljeg). Nju treba započeti pri prvoj
sumnji na ovu bolest, ne čekajući nalaze pretraga. Ako se dijeta
započne na vreme, simptomi se mogu postupno i povući. Dugoročna
prognoza lečene dece je dobra, iako ih dio može imati blagi
zaostatak u rastu, blaže govorne teškoće i druge diskretne mentalne
poremećaje. Bolesnici imaju povišene koncentracije galaktoze u
serumu i urinu. Žena koja zna da nosi gen za galaktozemiju mora
tokom trudnoće potpunosti prestati uzimati hranu koja sadrži
galaktozu. Galaktozemija se može u trudnoći sprečiti odgovarajućom
dijetom. Ukoliko majka ima visok nivo galaktoze u krvi, ona može
prolaziti kroz posteljicu i izazvati kataraktu. Osobe sa ovim
poremećajem moraju se odreći galaktoze za celi život [16].
Skrining na glutarnu aciduriju tip I
Glutarna acidurija tip 1 je teški nasledni neurometabolički
poremećaj čiji se klinički ishod poboljšao nakon primene programa
skrininga novorođenčadi i brzog početka presimptomatskog
metaboličkog lečenja. Glutarna acidemia tipa I je protip tzv.
cerebralnih organskih acidurija i rezultat je naslednog poremećaja u
metabolizmu aminokiselina lizina, hidroksilizina i triptofana, zbog
nedostatka mitohondrijskog enzima glutaril-CoA-dehidrogenaze. U
bolesnika s manjkom enzima nakupljaju se glutarična a u manjoj meri
3-OH-glutarična i glutakonična kiselina u mozgu [17]. Procenjena
prevalencija bolesti se kreće od 1:125,000 do 1:250 novorođenčadi u
genetski visokorizičnim populacijama [18]. Nelečena bolest najčešće
uzrokuje sliku akutnog oštećenja mozga s teškim
distoničko-diskinetičkim poremećajem. Bolest je asiptomatska do dobi
od obično pola godine do godinu dana kada se kod deteta u sklopu
neke infekcije, imunizacije ili druge stresne situacije razvije tzv.
encefalopatična kriza u kojoj stradaju bazalne ganglije.
Slika 4. Dete sa glutarnom acidurijom tip Ⅰ
Izvor:https://upload.wikimedia.org/wikipedia/commons/thumb/1/19/GA1_posture2.jpg/220px-GA1_posture2.jpg

Bolest se karakteriše neurorazvojnim poremećajima, uključujući:
kašnjenje ili deficit u razvoju govora, poteškoće u učenju,
poremećaj u intelektualnom razvoju, epilepsiju, makrocefaliju [19].
Kombinovana metabolička terapija uključuje ishranu sa niskim
sadržajem lizina, suplementaciju karnitinom i hitno lečenje sa
ciljem sprečavanja katabolizma i minimiziranja izloženosti CNS-a
lizinu i njegovim toksičnim metaboličkim nusproizvodima [20].
Skrining na cističnu fibrozu
Neonatalni skrining za cističnu fibrozu je optimizovao prognozu za
pacijente omogućavajući veoma ranu multidisciplinarnu negu. Tokom
proteklih 20 godina, programi skrininga su doživeli veliku
međunarodnu ekspanziju. Polovinom 20 veka, kada je bolest otkrivena,
deca oboljela od cistične fibroze umirala su tokom prve godine
života. Ranom dijagnostikom, poboljšanim lečenjem i primenom novih
lekova, prosečni životni vek obolelih je 40 godina. U zemljama koje
su uvele neonatalni skrinig, životni vek obolelih je značajno
produžen, poboljšan je kvalitet života obolelih i njihovih porodica.
Cistična fibroza je autozomno recesivna bolest koju karakteriše
insuficijencija pankreasa i hronična endobronhijalna infekcija
disajnih puteva. Hronična infekcija disajnih puteva dovodi do
progresivnih bronhiektazija i konačno respiratorne insuficijencije,
što je vodeći uzrok smrti kod pacijenata sa cističnom fibrozom.
Ostale komplikacije uključuju sinusitis, dijabetes melitus,
opstrukciju creva, hepatobilijarnu bolest, hiponatremijsku
dehidraciju i neplodnost [21]. Prednost ranog postavljanja dijagnoze
cistične fibroze neonatalnim skriningom je višestruka: primena
preventivnih i ranih terapijskih intervencija, redovno kontrolisanje
i rano otkrivanje komplikacija, značajno bolje preživljavanje
obolelih, duži i kvalitetniji život obolelih, sporija progresija
plućne bolesti, prevencija malnutricije, bolja uhranjenost,
omogućavanje normalnog rasta i razvoja dece.
ZAKLJUČAK
Dijagnostikovanje bolesti u najranijoj životnoj dobi omogućava
brzi terapijski pristup, koji dovodi do normalnog psihofizičkog
rasta i razvoja deteta i prevenira trajna telesna i intelektualna
oštećenja. Nasledne metaboličke i endokrinološke bolesti se
karakterišu visokim procentom telesnog i mentalnog invaliditeta,
koji pogađa ne samo zdravlje i socijalno funkcionisanje deteta već i
celu porodicu, zajednicu i društvo. Skrining na kongenitalnu
hipotireozu se počeo primenjivati u Crnoj Gori u 2007 godini. To je
jedino endokrinološko obolenje koje je predmet novorođenačkog
skrininga u Crnoj Gori.
LITERATURA
- Advisory Committe on Heritable Disorders in Newborn and
children; Recommended Uniform Screening Panel. [Internet]
[Citirano 2021 Novembar 02]. Dostupno na:
https://ww.hrsa.gov/advisory-committes/heritable
dosorders/rusp/index.html
- Stone WL, Basit H, Los E. Phenylketonuria. 2021 Nov 5. In:
StatPearls [Internet]. Treasure Island (FL): StatPearls
Publishing; 2022. [Citirano 2022 Avgust 01]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/30570999/
- Woolf LI, Adams J. The Early History of PKU. Int J Neonatal
Screen. 2020;6(3):59. [Citirano 2022 Jul 28]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/33239585/
- Mancilla VJ, Mann AE, Zhang Y, Allen MS. The Adult
Phenylketonuria (PKU) Gut Microbiome. Microorganisms.
2021;9(3):530. [Citirano 2022 Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/33806544/
- Wiedemann A, Oussalah A, Jeannesson É, Guéant JL, Feillet F.
La phénylcétonurie - De la diététique à la thérapie génique
[Phenylketonuria, from diet to gene therapy]. Med Sci (Paris).
2020;36(8-9):725-734. [Citirano 2022 Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/32821049/
- van Spronsen FJ, Blau N, Harding C, Burlina A, Longo N,
Bosch AM. Phenylketonuria. Nat Rev Dis Primers. 202;7(1):36.
[Citirano 2022 Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/34017006/
- Chen S, Zhu M, Hao Y, Feng J, Zhang Y. Effect of Delayed
Diagnosis of Phenylketonuria With Imaging Findings of Bilateral
Diffuse Symmetric White Matter Lesions: A Case Report and
Literature Review. Front Neurol. 2019;10:1040. [Citirano 2022
Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/31636599/
- MacDonald A, van Wegberg AMJ, Ahring K, Beblo S,
Bélanger-Quintana A, Burlinaet et al., APKU dietary handbook to
accompany PKU guidelines. Orphanet J Rare Dis. 2020;15(1):171.
[Citirano 2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/32605583/
- Arduini GAO, Balarin MAS, Silva-Grecco RLD, Marqui ABT.
KNOWLEDGE OF PUERPERAL MOTHERS ABOUT THE GUTHRIE TEST. Rev Paul
Pediatr. 2017;35(2):151-157. [Citirano 2022 Avgust 05]. Dostupno
na: https://pubmed.ncbi.nlm.nih.gov/28977324/
- Guerri G, Bressan S, Sartori M, Costantini A, Benedetti S,
Agostini F et al., Hypothyroidism and hyperthyroidism. Acta
Biomed. 2019;90(10-S):83-86. [Citirano 2022 Avgust 05]. Dostupno
na: https://pubmed.ncbi.nlm.nih.gov/31577260/
- American thyroid association (2020). A review of the 2020
guidlines for congenital hypothyroidism. [Citirano 2021 Novembar
03] Dostupno na: thyroid.org/congenital-hypothyroidism
- British Thyroid foundation (2018). Congenital
hypothyroidism. [Citirano 2021 Novembar 02] Dostupno na
:www.british-thyroid-association.org
- Yuzyuk T, Balakrishnan B, Schwarz EL, De Biase I, Hobert J,
Longo N et al., Effect of genotype on galactose-1-phosphate in
classic galactosemia patients. Mol Genet Metab. 2018
Nov;125(3):258-265. [Citirano 2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/30172461/
- Lak R, Yazdizadeh B, Davari M, Nouhi M, Kelishadi R. Newborn
screening for galactosaemia. Cochrane Database Syst Rev.
2017;12(12):CD012272. [Citirano 2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/29274129/
- Hrvatski liječnički zbor u saradnji sa farmaceutskom tvrtkom
MSD (2014) MSD priručnik dijagnostike i terapije. [Citirano 2022
Januar 06]. Dostupno na:
http://www.msd-prirucnici.placebo.hr/msd-prirucnik/pedijatrija/nasljedne-metaboličke-bolesti/galaktozemija
- Kiss E, Balogh L, Reismann P. Klasszikus galactosaemia
dietetikai kezelési lehetőségei [Diet treatment of classical
galactosemia]. Orv Hetil. 2017;158(47):1864-1867. Hungarian.
[Citirano 2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/29153024/
- Boy N, Mohr A, Garbade SF, Freisinger P, Heringer-Seifert J,
Seitz A et al., Subdural hematoma in glutaric aciduria type 1:
High excreters are prone to incidental SDH despite newborn
screening. J Inherit Metab Dis. 2021;44(6):1343-1352. [Citirano
2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/34515344/
- Boy N, Mengler K, Heringer-Seifert J, Hoffmann GF, Garbade
SF, Kölker S. Impact of newborn screening and quality of therapy
on the neurological outcome in glutaric aciduria type 1: a
meta-analysis. Genet Med. 2021;23(1):13-21. doi:
10.1038/s41436-020-00971-4. Epub 2020 Sep 28. PMID: 32981931;
PMCID: PMC7790745.
- Pokora P, Jezela-Stanek A, Różdżyńska-Świątkowska A,
Jurkiewicz E, Bogdańska A, Szymańska E, Rokicki D, Ciara E,
Rydzanicz M, Stawiński P, Płoski R, Tylki-Szymańska A. Mild
phenotype of glutaric aciduria type 1 in polish patients - novel
data from a group of 13 cases. Metab Brain Dis.
2019;34(2):641-649. doi: 10.1007/s11011-018-0357-5. Epub 2018
Dec 20. PMID: 30570710; PMCID: PMC6428789.
- Larson A, Goodman S. Glutaric Acidemia Type 1.. In: Adam MP,
Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa
GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA):
University of Washington, Seattle; 2019;1993–2022. PMID:
31536184.
- Goetz D, Ren CL. Review of Cystic Fibrosis. Pediatr Ann.
2019;48(4):e154-e161. doi: 10.3928/19382359-20190327-01. PMID:
30986316.
|
|
|
|
